
The aim of the “Sentinel” national surveillance program is to monitor the circulating respiratory viruses, including SARS-CoV-2 variants, and hence underpin public health actions.
In week 24/2021, the variant B.1.617.2 became the dominant one in Luxembourg with an overall frequency of 59,4% (CI 47,4% – 71,4%, p<0,05), while the B.1.1.7 variant frequency dropped to 29,7% (CI 18,5% – 40,9%, p<0,05). One case of SARS-CoV-2 B.1.351 (1,5%, CI 0% – 4,5%, p<0,05) and no case of the P.1 variant were detected.
The representative sample size was estimated, based on the number of positive cases in Luxembourg for week 24 (90). The minimum sample size required to detect prevalence of B.1.617.2 (34%) reported in week 23, with an error margin of 5%, was estimated to be 72 specimens. This number corresponds to a coverage of 80 %, which exceeds the minimum coverage recommended by ECDC (10%). The sequencing results of week 24 are not representative of the circulating variants in Luxembourg with an error margin of 5%.
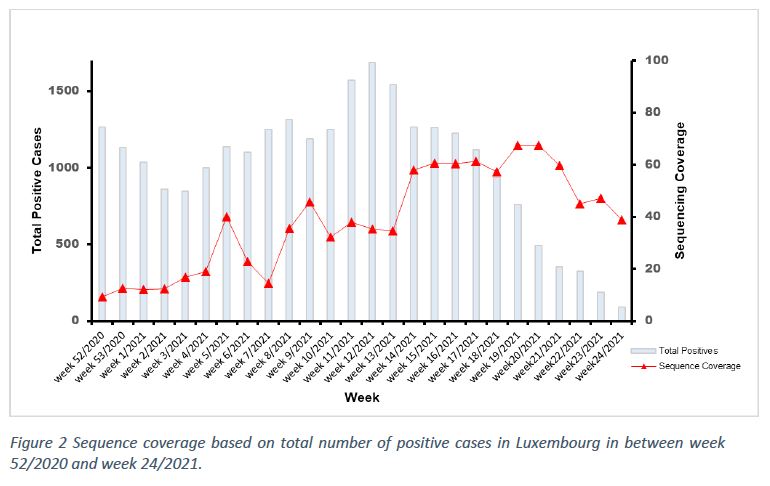
The total number of sequences performed this week was 284, with 64 specimens having been collected in the time frame of week 24/2021. The sequencing coverage – based on sequenced samples collected in week 24 and corresponding to Luxembourg residents – was 66,7% of all positive cases in Luxembourg.

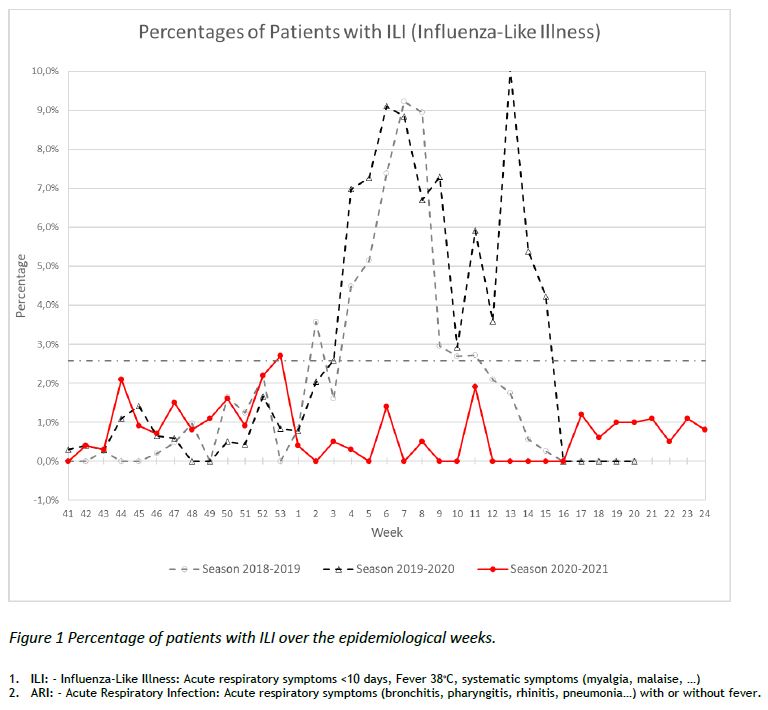
The “Sentinel” surveillance network reported 374 consultations in week 24 (14/JUNE/2021 – 20/JUNE/2021). There were 3 cases of ILI1, corresponding to 0,8% of the consultations, as shown in Figure 1. The number of consultations for ARI2 was 37, which represents 9,9% of the consultations.
Covid Consultation Centres have been closed since 16/May/2021. We are currently working on an alternative in partnership with private laboratories.
The National Reference Laboratory for Acute Respiratory Infections at LNS continues to improve the representativeness of the pool of sequenced specimens to reach real-time epidemiology, by implementing the following weekly sequencing activities:
The representative sequencing sample was based on the minimum number of specimens required to extrapolate prevalence of VOC variants with error rate of 5%. The representative sample was estimated based on the number of positive cases in Luxembourg in week 24 (90). The minimum sample size required to detect prevalence of B.1.617.2 (34%) reported in week 23 with an error margin of 5% was estimated to be 72 specimens. The calculation was based on a sample size calculation tool that uses the expected prevalence of the variant in the total population. (Population Proportion – Sample Size – Select Statistical Consultants (select-statistics.co.uk). This number represented a coverage of 80%, which exceeds the minimum coverage recommended by ECDC (10%). The number of non-targeted specimens from Luxembourgish residents sequenced this week was 35. Therefore, our sequencing results this week are not representative of the circulating variants in Luxembourg.
The starting material used for sequencing is respiratory specimens (nasopharyngeal or oropharyngeal swabs) that have already been tested positive by RT PCR.
The LNS sequencing data sharing strategy includes sharing of the sequencing data with GISAID EpiCov database (www.gisaid.org ) on a periodic basis.
Last week the microbial genomics platform at the LNS sequenced 284 specimens, with 64 collected in week 24/2021. The sequencing pool referring to Luxembourgish residents represents 66,7% of new infections reported in Luxembourg in week 24/2021. Among these 68 specimens, 25 specimens were reported to be part of a cluster or outbreak investigation, and 6 specimens were from non-residents (2 specimen overlapping). This leads to 35 specimens, collected in week 24, and not being a representative population sequencing sample. In the population sample of residents, the frequency of B.1.617.2 was 54,3%, while the frequency for B.1.1.7 was 34,3%. One case has been detected for the B.1.351 variant, representing 2,9%.
The population sequencing coverage in week 24/2021 was 38,9% (Figure 2). Based on statistical inference, the frequency of the reported variants in week 24/2021 is not representative of the circulating variants in Luxembourg with a margin of error of 5%.
Lineages (variants) have been assigned based on Rambaut et al by means of Phylogenetic Assignment of Named Global Outbeak LINeages (pangolin) software (v3.0.3, pangoLEARN version 2021-05-27).
The lineage nomenclature system that we use is the one proposed by Rambaut et al. that focuses on actively circulating virus lineages (https://cov-lineages.org ).
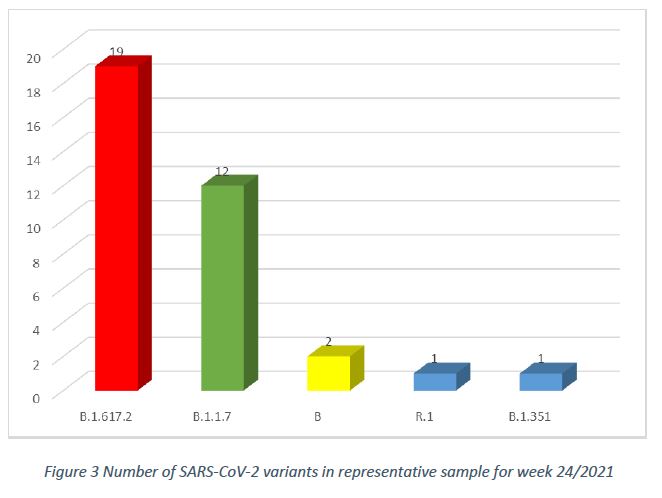
In week 24/2021, in the population sample, after removal of cluster samples, and excluding specimens collected from non-residents, there were 5 circulating SARS-CoV-2 variants, with the main three variants being B.1.617.2 (54,3%, CI 37,8% – 70,8%), B.1.1.7 (34,3%, CI 18,6% – 50%) and B (5,7%, CI 0% – 13,4%), as shown in Figure 3.
Among specimens collected within the week 24/2021, 19 cases of the B.1.1.7 variant have been detected, representing 29.7% of the specimens in the week’s sequencing pool (by comparison, the week 23/2021 pool had shown a frequency of 51.6% of this variant, including additional specimens having been sequenced from previous weeks). The total case count of sequenced variant B.1.1.7 (Alpha) was 6696 by week 24/2021.
In the collection period of week 24/2021, 1 case of the South African variant B.1.351(Bêta) has been detected, representing 1.6% (by comparison, the week 23/2021 pool had shown 0.8% of this variant). The total case count of sequenced variant B.1.351 was 1123 by week 24/2021.
No new case of the P.1 variant has been detected in week 24/2021.The total case count of sequenced variant P.1(Gamma) was 126 by week 24/2021 (including additional sequencing of previous weeks).
In week 24/2021, no new case of B.1.525 has been detected. The case count by week 24 for B.1.525 is 48 (including additional sequencing of previous weeks).
In week 24/2021, 38 additional cases of the Delta variant B.1.617.2 have been detected (latest sampling date 20/JUNE/2021). The case count by week 24 for B.1.617.1 increases by retrospective sequencing to 9, and for B.1.617.2 it raises to 231. Since May 6th, B.1.617.2 has been escalated by Public Health England from a variant under investigation to a variant of concern (Figure 4).
Lineage B.1.1.7 is characterized by several spike protein mutations, including N501Y, H69/V70del and P861H. The variant seems to have a considerable epidemiological impact, as it has a higher transmissibility rate.
Lineage B.1.351 holds numerous spike protein mutations, of which three are located in the receptor binding domain (K417N, E484K and N501Y), and are therefore relevant for antibody binding. Similarly to B.1.1.7, a higher transmissibility rate and viral loads seem to be associated with this variant. Due to the K417N and E484K mutations, an impact on vaccination efficacy and possibility of reinfection is subject to scientific investigation.
Lineage P.1 (descendent of B.1.1.28), initially found in the Amazon region, has a similar mutation profile as the South African variant, including E484K and N501Y. Concerns are, as for the South African variant, higher transmissibility and a decreased protection by neutralizing antibodies.
Lineage B.1.525 carries several mutations of biological significance, including E484K, Q677H and F888L. It does not carry N501Y, but a set of deletions similar to the B.1.1.7 variant.
Lineage B.1.617 is a variant first detected in India and was designated “Under Investigation” on 1st April 2021 by Public Health England. It contains a number of spike mutations associated with antigenic escape or found in other variants of concern, including L452R, E484Q and P681R. Subtype B.1.617.2 does not carry S:E484Q and seems to be more transmissible, as B.1.1.7 (increasing confidence). Neutralization studies show reductions in cross-neutralizing activity between B.1.1.7 and B.1.351.

By week 24/2021, 90.6 % of the variants detected in the sequenced specimen pool are declared as Variants of Concern, including the B.1.617.2, B.1.1.7 and B.1.351 variants.
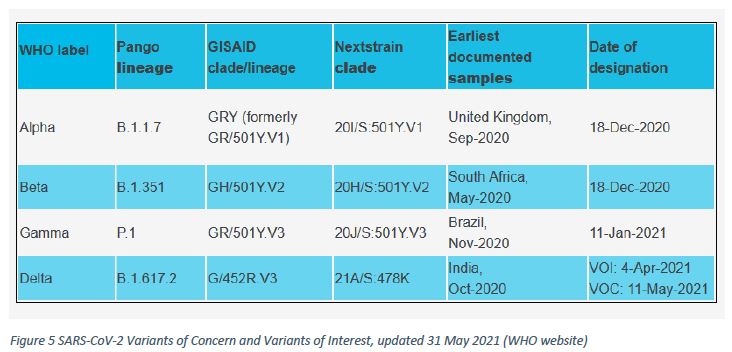
The ReViLux will continue to use the (Pango) system to allow easier visualisation of links between any evolving variants and their ancestor (Figure 5).
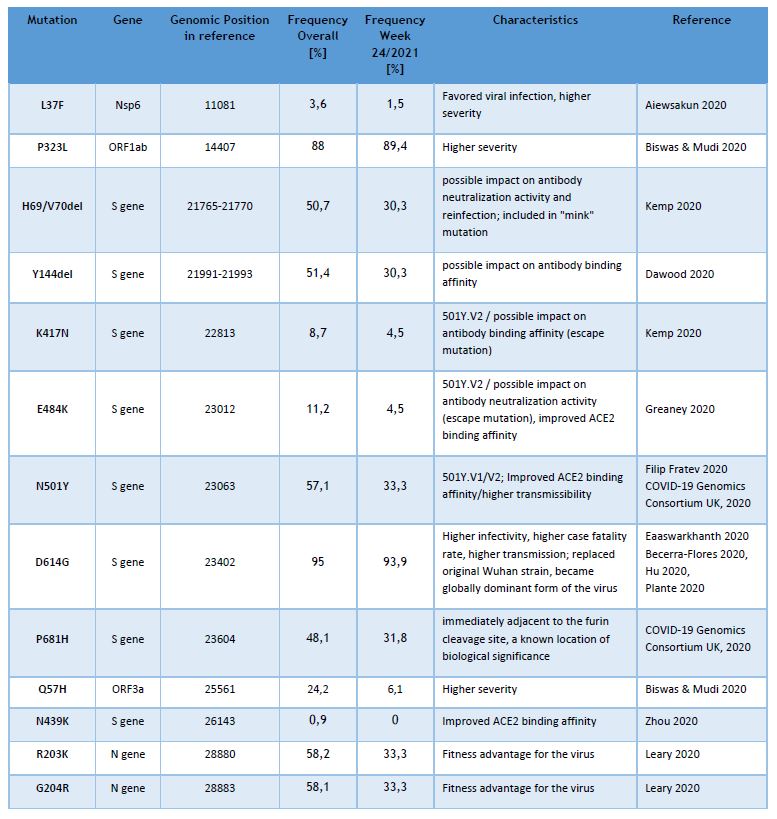
Currently the LNS genomic surveillance program – independently from lineage calling – notes the occurrence of 13 different known SARS-CoV-2 mutations, assumed to have clinical and epidemiological relevance. The list of observed mutations is being updated continually, based on the appearance and prevalence of SARS-CoV-2 variants.
The following table provides the overall frequencies of these mutations, detected in the lineage-assignable genome sequences, analyzed between 01/SEP/2020 and 20/JUNE/2021 (N=13149), as well as the frequencies in week 24/2021.
Genomic sequencing of SARS-CoV-2. A guide to implementation for maximum impact on public health. WHO, 8 January 2021.
COVID-19 data portal. 2020 (https://www.covid19dataportal.org/sequences )
J Hadfield et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018;34:4121-4123
A Rambaut et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 2020;5:1403-1407
https://github.com/cov-lineages/pangolin
For more information on lineages visit: https://cov-lineages.org
For more information and statistics on Covid-19 infections in Luxembourg visit: https://covid19.public.lu/en.html