
The aim of the “Sentinel” national surveillance program is to monitor the circulating respiratory viruses, including SARS-CoV-2 variants, and hence underpin public health actions. ECDC and WHO have recently recommended that national sequencing programs should cover a minimum of 10% of all SARS-CoV-2 positive specimens, or 500 specimens, whichever is less.
In week 5/2021, the SARS-CoV-2 sequencing coverage had reached 27% of all identified positive cases, yielding a representative population sequencing sample. Luxembourg reported the second highest sequencing coverage in EU, surpassed only by Denmark. In week 6/2021, this coverage was 19.5%.
In week 6/2021, the overall prevalence of the SARS-CoV-2 B.1.1.7 variant in the sequencing sample was 53%. After correction for the bias linked to sequencing of targeted, instead of random specimens, the prevalence of the B.1.1.7 variant, in the representative population sequencing sample, was 57%. A circumscript outbreak of 17 cases of the B.1.351 variant, appeared in the hospital environment.

At present, the scope of the ReViLux report is to provide (i) surveillance and descriptive epidemiology data, including data on molecular phylogenies (identification of importation events, changes in outbreak size over time), and (ii) phylogenetic interpretation of regional virus spread and circulation in Luxembourg, and cluster investigation. The scope of the ReViLux report is not to attempt phylogeographical reconstructions in Luxembourg, as a whole.
The “Sentinel” surveillance network is composed of 37 general practicioners and paediatricians around Luxembourg (General Practice and Paediatrics). This week we had 145 consultations reported by 9 of these general paractitioners and pediatricians (24% of the network size).
During week 6 (8/2/2021 – 14/2/2021), there were 2 cases of ILI1 representing 1.37% of consultations (epidemiological threshold is 2 new cases per week), as shown in Figure 1. The percentage of consultations for ARI2 was 14%.
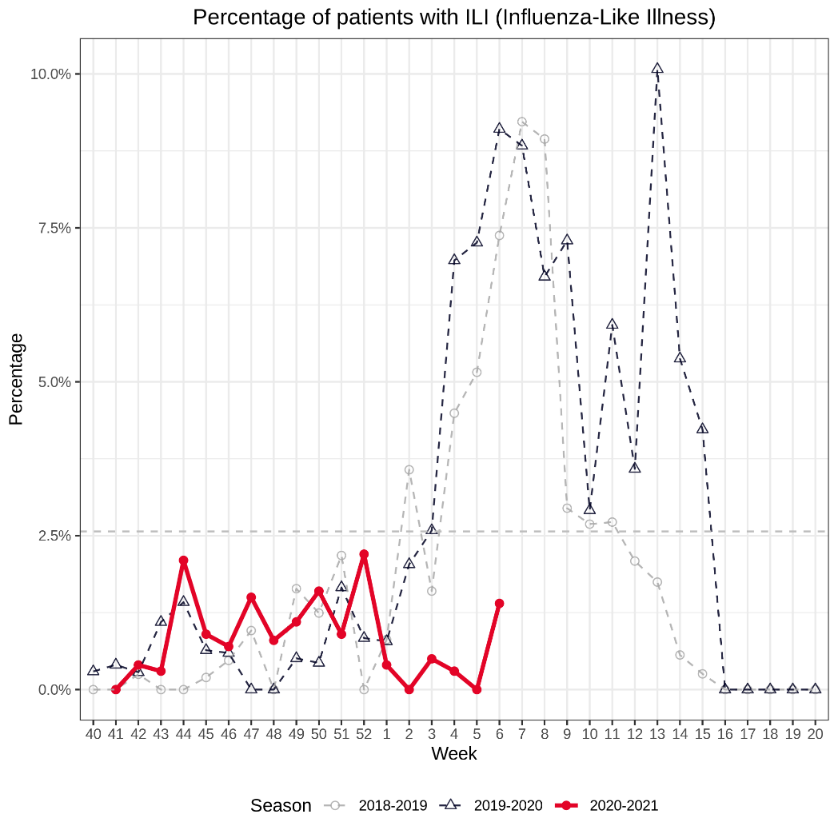
Figure 1 Percentage of patients with ILI over the epidemiological weeks
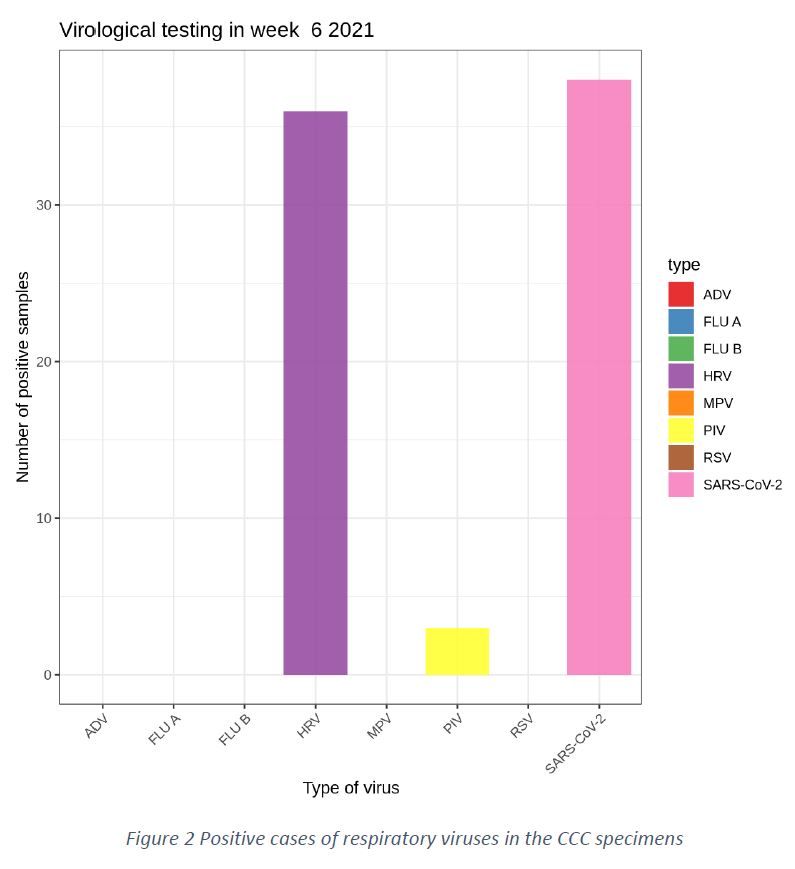
Patients presenting with ILI and ARI at the Covid Consultation Centre (CCC) in Luxembourg were tested using a respiratory virus panel (ADV = Adenovirus, FLU A = Influenza A, FLU B = Influenza B, HRV = Human Rhinovirus, MPV = Human metapneumovirus, PIV = Parainfluenza virus, RSV = Respiratory Syncytial Virus, SARS-CoV-2 = Severe acute respiratory syndrome coronavirus 2). The SAR-COV-2 was the most prevalent respiratopry virus detected in the “Sentinel” network, with 38 positive cases (20.8%). The wave of Human Rhinovirus (HRV), circulating currently in Luxembourg, continued in week 6/2021, with 36 positive cases in 183 tests (19.7%). Only three positive cases of Parainfluenza viruses were detected. No cases of Influenza A/B were detected, indicating absence of circulation of Influenza viruses in Luxembourg as shown in Figure 2.
In Luxembourg, we have tested 183 samples from the Sentinel surveillance network, as compared to 1268 samples tested in Europe, in the week 06/2021. None of these 1268 specimens tested for influenza viruses were positive. The influenza epidemic in the European Region has usually reached its peak by this point of the year but, despite widespread and regular testing for influenza, reported influenza activity still remains at a very low level, likely due to the impact of the various public health and social measures, implemented to reduce transmission of SARS-CoV-2 (Source: FluNews Europe).
The National Reference Laboratory for Acute Respiratory Infections at LNS continues to improve the representativeness of the pool of sequenced specimens to reach real-time epidemiology, by implementing the following weekly sequencing activities:
The starting material used for sequencing is respiratory specimens (nasopharyngeal or oropharyngeal swabs) that have already been tested positive by RT PCR.
Last week the microbial genomics platform at the LNS has sequenced 390 specimens. Out of these, 214 specimens were collected in week 6/2021. This represents 19.5% of new infections reported in Luxembourg in week 6/2021. Among the 214 specimens, 38% (82 samples) corresponded to targeted specimens (contact tracing with 46 specimens and cluster investigation with 36 specimens).
After filtering out specimens referred for sequencing in a targeted way, the population sequencing coverage in week 6/2021 was 12%. This exceeds the ECDC recommendation of 10% being the minimum requirement for a representative population sequencing sample.
Lineages (variants) have been assigned based on Rambaut et al by means of Phylogenetic Assignment of Named Global Outbeak LINeages (pangolin) software (v2.3, pangoLEARN version 2021-02-18).
The lineage nomenclature system that we use is the one proposed by Rambaut et al. that focuses on actively circulating virus lineages (https://cov-lineages.org).
In the sampling period of week 6/2021, in the population representative sample (132 specimens), 13 variants were detected, with the main three variants being B.1.1.7 (57.6%), B.1.160 (13.6%), followed by B.1.221 (7.6%), as shown in Figure 3.
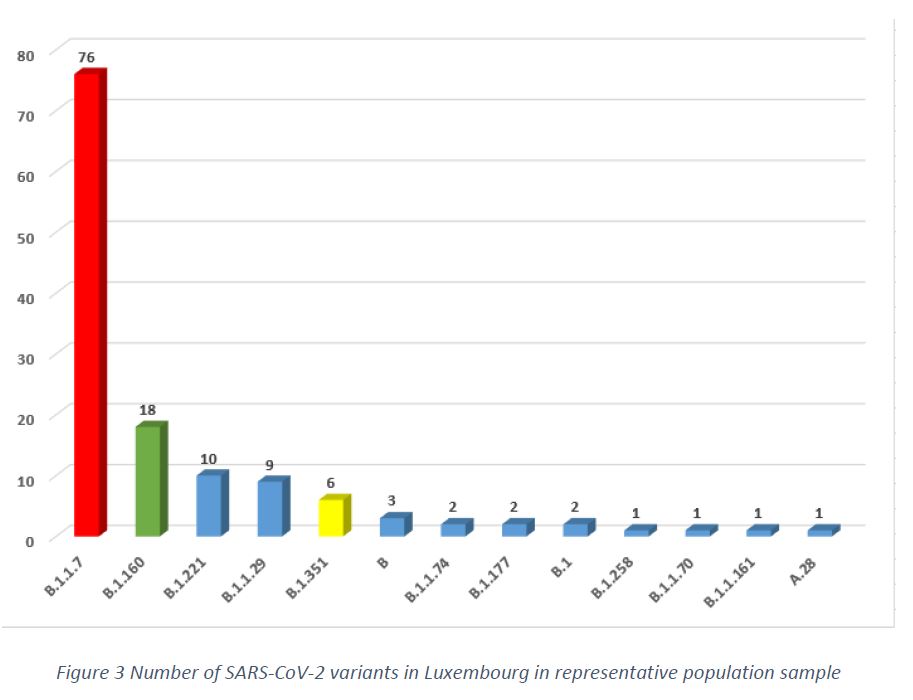
Among specimens collected within the week 6/2021, 114 cases of the B.1.1.7 variant have been detected, representing 53,5% of the specimens in the week’s sequencing pool (by comparison, the week 5/2021 pool, which also included specimens from previous weeks’ collections, had shown 54,2% of this variant). The total case count of sequenced variant B.1.1.7 was 403 by week 6/2021. The earliest collection date for this variant remains 19/DEC/2020 and the latest is 14/FEB/2021.
In the collection period of week 6/2021, 24 cases of the South African variant B.1.351 have been detected. The total case count of sequenced variant B.1.351 was 52 by week 6/2021. The earliest collection date for this variant remains 11/JAN/2021 and the latest is 14/FEB/2021.
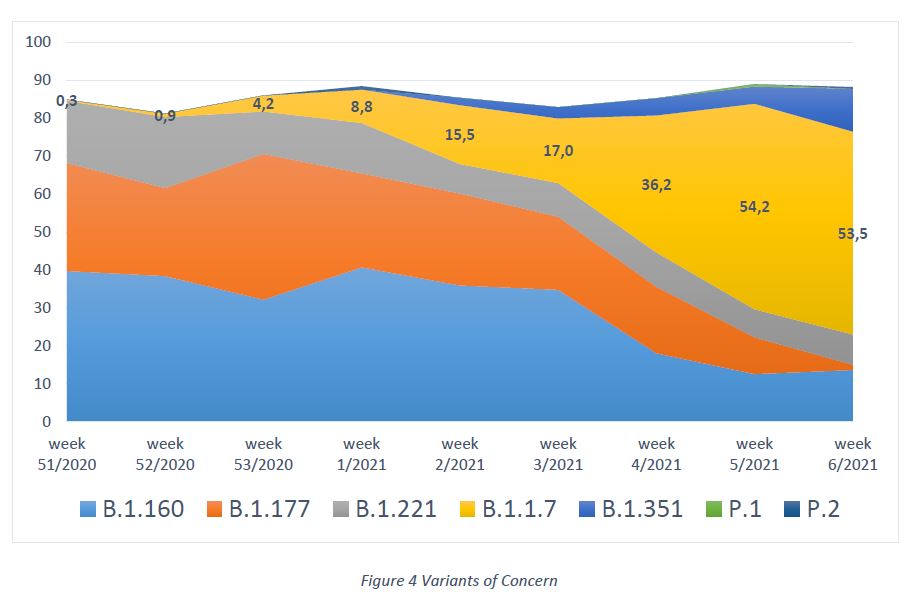
In week 6/2021, no additional cases of the Brazilian variant P.1 were detected, therefore the total case count of variant P.1 was 1 by week 6/2021 (collection date 02/FEB/2021). Last week, the pangolin group, which provides a global report on novel coronavirus haplotypes, published two additional variants of concern, assigned as A.23.1 and B.1.525. Both variants have already been reported by neighboring countries (Belgium, France). However, none of these variants has been detected in Luxembourg (Figure 4).
Lineage B.1.1.7 is characterized by several spike protein mutations, including N501Y, H69/V70del and P861H. The variant seems to have a considerable epidemiological impact, as it has a higher transmissibility rate.
Lineage B.1.351 holds numerous spike protein mutations, of which three are located in the receptor binding domain (K417N, E484K and N501Y), and are therefore relevant for antibody binding. As for B.1.1.7, a higher transmissibility rate and viral loads seem to be associated with this variant. Due to the K417N and E484K mutations, an impact on vaccination efficacy and possibility of reinfection is subject to scientific investigation.
Lineage P.1 (descendent of B.1.1.28), initially found in the Amazon region, has a similar mutation profile as the South African variant, including E484K and N501Y. Concerns are, as for the South African variant, higher transmissibility and a decreased protection by neutralizing antibodies.
Of note, additional specimens are in the pipeline for sequencing, so that numbers may change with increasing representativeness of circulating variants and proportions.
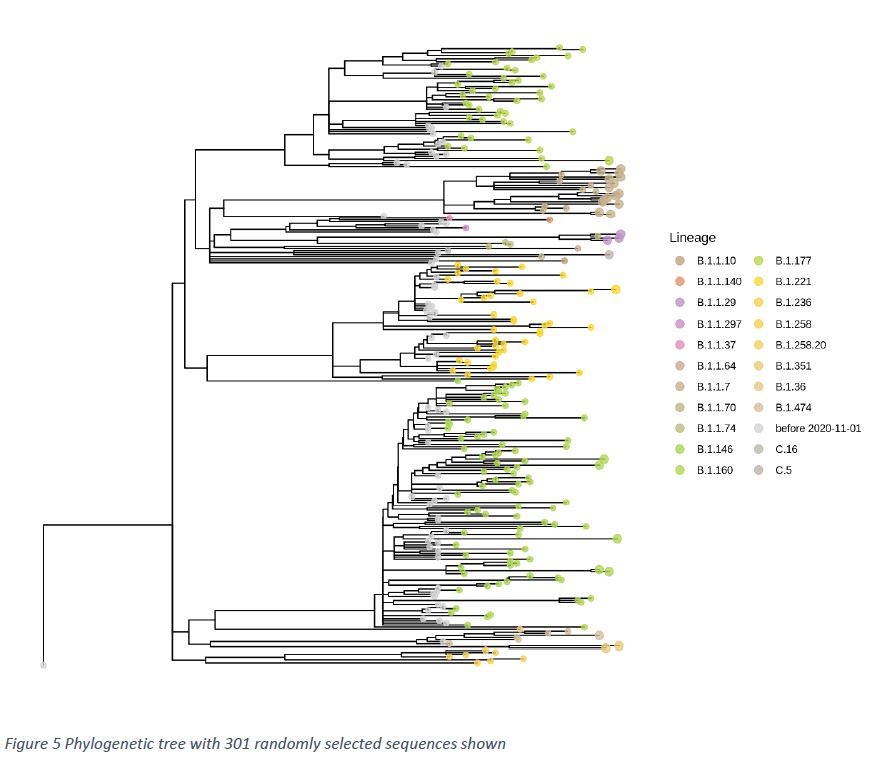
Phylogenetic analysis is done by means of Nextstrain (https://nextstrain.org/sars-cov-2/) and is based on genetic distances including a timeline on the x-axis. The phylogenetic tree is rooted against the first sequence obtained in Luxembourg (29/FEB/2020), which is considered as our “outgroup”. Sequences obtained before 01/NOV/2020 are shown as grey dots. Sequences obtained within the last 2 weeks, are represented in colour and by greater dot diameter. In the context of increasing numbers of sequences, a random subsampling of 300 sequences over the whole sequencing period, starting 01/SEP/2020, was performed.
The phylogenetic tree shows four visually well separated clusters. These clusters correspond to the lineages B.1.160, B.1.177, B.1.1.7 and B.1.221. Due to random selection of sequences and the intentional insertion of variants of concern, the cluster representation is mostly qualitative and does not necessarily correlate with relative prevalence of the respective variants (Figure 5).
Lineage B.1.1.7 (represented by light brownish dots) forms a well distinguishable cluster, due to the high number of accumulated mutations. B.1.1.7 subtrees might indicate different subgroups, possibly related to different routes of infection.
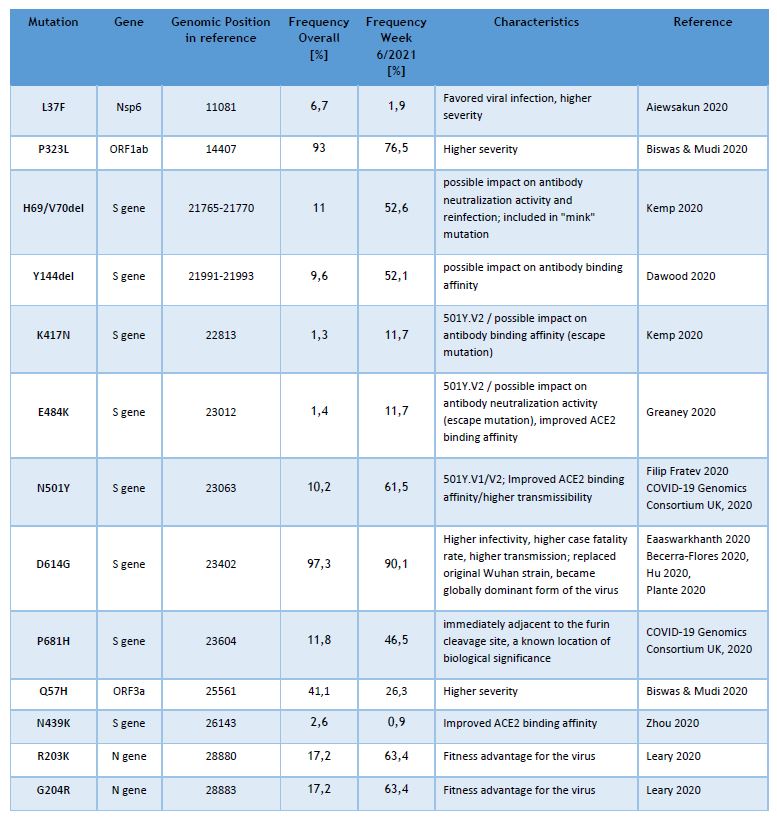
Currently the LNS genomic surveillance program – independently from lineage calling – notes the occurrence of 13 different known SARS-CoV-2 mutations, assumed to have clinical and epidemiological relevance. The list of observed mutations is being updated continually, based on the appearance and prevalence of SARS-CoV-2 variants.
The following table provides the overall frequencies of these mutations, detected in the lineage-assignable genome sequences, analyzed between 01/SEP/2020 and 14/FEB/2021 (N=4392), as well as the frequencies in week 6/2021.
The ReViLux data are communicated as support to the understanding of respiratory virus transmission dynamics, including introduction of new variants, to the evaluation of the impact of response measures, to the contact tracing and the investigation of clusters.
The SARS-CoV-2 sequencing program at the LNS continues to expand, both with higher coverage and with retrospective epidemiological metadata annotations.
Genomic sequencing of SARS-CoV-2. A guide to implementation for maximum impact on public health. WHO, 8 January 2021.
COVID-19 data portal. 2020 (https://www.covid19dataportal.org/sequences )
J Hadfield et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018;34:4121-4123
A Rambaut et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 2020;5:1403-1407
https://github.com/cov-lineages/pangolin
Y Guangchuang et al. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution 2017;8:28-36
For more information on lineages visit: https://cov-lineages.org
For more information and statistics on Covid-19 infections in Luxembourg visit: https://covid19.public.lu/en.html