2024 07-23
- The Laboratory
- Organization
- Departments
- Jobs
- Analysis book
- Contact
- News
- Publications
- Download

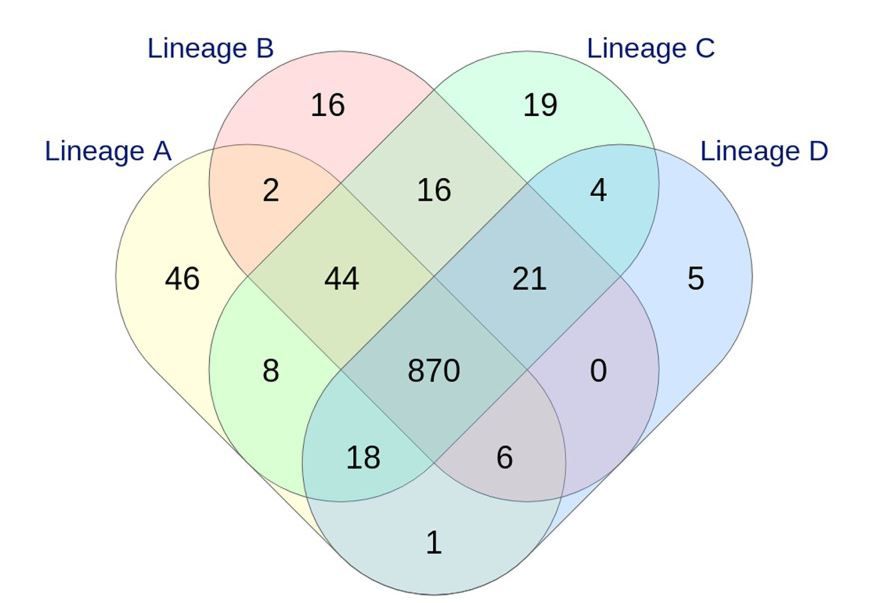
According to the World Health Organization, Campylobacter spp. is one of four key global causes of bacterial diarrheal diseases around the globe with C. jejuni species involved in more than 80% of the human infection cases. The monitoring of C. jejuni genetic profiles circulating in Luxembourg was initiated 15 years ago. Molecular comparison of isolates, initially based on an extended MLST Sanger-based method, revealed an unexpected endemic pattern in the temporal distribution of human genotypes. This is one of the many research projects of the microbial epidemiology and genomics team of the LNS microbiology department is working on.
The team initially aimed to investigate the occurrence of stable lineages by implementing whole genome sequencing (WGS), associated with comprehensive and internationally validated genotyping schemes. In this pilot study, four particular lineages were investigated due to the high number of corresponding strains in human infections and their occurrence in non-human samples (animals, food, surface waters). Results indicated a good agreement between three cgMLST (core genome Multi-Locus Sequence-Typing) schemes tested, suggesting that isolates with the same extended-MLST profile (assessed by an in-house fingerprint method) shared the same core genome. Whatever the WGS-based typing scheme applied, epidemic or endemic signals were clearly detected. These findings support the clonal expansion of stable genomes in Campylobacter populations, exhibiting a multi-host profile and accounting for the majority of clinical human strains isolated for more than a decade in Luxembourg. Such recurring genotypes suggest persistence in environmental reservoirs, emphasizing the need to investigate their survival strategy in greater detail.
This is the research question of the thesis project of Morgane Nennig at LNS.
Link to the article: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7819963/